


UMSOM Researchers Identify Genetic Defect Linked to ALS
Researchers at the University of Maryland School of Medicine (UMSOM) have identified how certain gene mutations cause amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease. The pathway identified by the researchers may also be responsible for a certain form of dementia related to ALS. The finding could offer potential new approaches for treating this devastating condition, which causes progressive, fatal paralysis and sometimes mental deterioration similar to Alzheimer’s disease. Their discovery was published this week in the Proceedings of the National Academy of Sciences (PNAS) and included collaborators from Harvard University, University of Auckland, King’s College London, and Northwestern University.
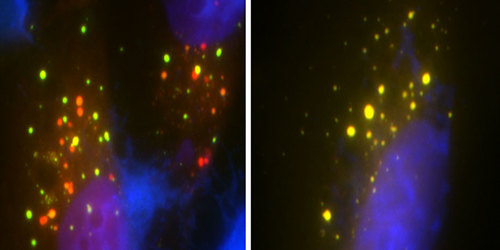
Cells on the left have the normal UBQLN2 gene and the red dots show the cell “garbage removal” process in action. The cells on the right have gene mutations that disrupt this process allowing toxic matter to build up in cells.
More than 5,000 Americans are diagnosed with ALS every year, a condition that is usually fatal and has no cure. Patients with ALS slowly lose the ability to move their muscles, leading to problems with basic functions such as breathing and swallowing. About half of ALS patients also develop dementia. Genetic studies of families with a predisposition to develop ALS have shown that the condition can be associated with certain gene mutations. Some of these mutations involve the gene UBQLN2 which regulates the disposal of misfolded “garbage” from the body’s cells. Until now, researchers did not fully understand how UBQLN2 mutations interfere with this pathway and cause ALS.
“We mapped out the process by which ubiquilin-2 (UBQLN2) gene mutations disrupt an important recycling pathway that cells use to get rid of their trash,” said Mervyn Monteiro, PhD, professor of anatomy and neurobiology, who is affiliated with the UMSOM’s Center for Biomedical Engineering and Technology (BioMET). “Without this recycling, misfolded proteins build up in the nerve cell and become toxic, eventually destroying the cell. This destruction could lead to neurodegenerative disorders like ALS.”
To investigate how UBQLN2 mutations cause ALS, Monteiro’s group used both human cells and UBQLN2-mutant mouse models for their investigations. The mouse models, which they described in a 2016 PNAS publication, mimic the progression of the disease in people who inherit these gene mutations.
Monteiro’s group first removed the UBQLN2 gene from human cells and found it completely stalled the recycling pathway. They then reintroduced either the normal gene or one of five gene mutations into the cells. They found that reintroduction of normal UBQLN2 restored the recycling pathway while all five of the gene mutations failed to restart the pathway.
Using the mouse model, Monteiro and his colleagues outlined the reason for the pathway disruption in the presence of gene mutations. They found that the mice with the gene mutations had reduced levels of a certain protein called ATP6v1g1, which is an essential part of a pump that acidifies the cell’s trash container in order to initiate the breakdown and recycling process.
“Our new findings are exciting because similar acidification defects have been found in Alzheimer’s, Parkinson’s and Down syndrome,” Monteiro said. “This suggests that restoration of the defect could have broad implications for not only treating ALS, but possibly other neurodegenerative diseases as well.”
The research study was supported by grants from the Packard Center for ALS Research at Johns Hopkins, the ALS Association, and the National Institutes of Health (grant number: R01-NS100008).
“The BioMET research team led by Monteiro continues to make important advances in understanding the mechanisms that give rise to ALS,” said Dean E. Albert Reece, MD, PhD, MBA, who is also executive vice president for medical affairs, University of Maryland, Baltimore, and the John Z. and Akiko K. Bowers Distinguished Professor, UMSOM. “Future treatments and preventive measures for this devastating disease would not be possible without this foundational work.”
